- ІМПЛЕМЕНТАЦІЙНИЙ РЕГЛАМЕНТ КОМІСІЇ (ЄС) 2018/150 від 30 сі…
- Стаття 1
- "Стаття 60a Спеціальні положення щодо перевірок, пов'язани…
- Стаття 2
- Стаття 3
- ДОДАТОК
- До додатків до Імплементаційного регламенту (ЄС) № 2016/12…
- Доповнення I
- Доповнення II
- Доповнення III
- Умови градієнта для оптимізації хроматографії
- ДОДАТОК VI
- ДОДАТОК VII
- ДОДАТОК VIII
- Доповнення
- ДОДАТОК IX
- Доповнення

Увійдіть, щоб зручно організувати та зберігати закони і судові рішення. Це безкоштовно.
Приєднуйтесь.
Зберігайте закони у приватних списках для швидкого доступу. Діліться публічними списками з іншими.
(До Розділу V: Економічне та галузеве співробітництво
Глава 17. Сільське господарство та розвиток сільських територій)
ІМПЛЕМЕНТАЦІЙНИЙ РЕГЛАМЕНТ КОМІСІЇ (ЄС) 2018/150
від 30 січня 2018 року
про внесення змін до Імплементаційного регламенту (ЄС) 2016/1240 стосовно методів дослідження та оцінювання якості молока і молочних продуктів, що підпадають під дію державних інтервенцій та відповідають критеріям для надання допомоги на зберігання на складах приватного користування
Беручи до уваги Договір про функціонування Європейського Союзу,
Беручи до уваги Регламент Європейського Парламенту і Ради (ЄС) № 1306/2013 від 17 грудня 2013 року про фінансування, управління і моніторинг спільної сільськогосподарської політики та про скасування регламентів Ради (ЄЕС) № 352/78, (ЄС) № 165/94, (ЄС) № 2799/98, (ЄС) № 814/2000, (ЄС) № 1290/2005 і (ЄС) № 485/2008 (-1), зокрема його статтю 62(2)(i),
(1) Делегований регламент Комісії (ЄС) 2016/1238 (-2) та Імплементаційний регламент Комісії (ЄС) 2016/1240 (-3) установлюють правила щодо застосування державних інтервенцій та допомоги на зберігання на складах приватного користування. Регламент Комісії (ЄС) № 273/2008 (-4) визначає методи, які слід застосовувати, щоб оцінити, чи молоко і молочні продукти відповідають вимогам допустимості, установленим у згаданих регламентах щодо державних інтервенцій та надання допомоги з метою зберігання на складах приватного користування.
(2) У світлі технічних розробок у методології, використовуваної для дослідження та оцінювання якості молока і молочних продуктів, необхідно внести істотні зміни з метою спрощення та оновлення покликань на стандарти ISO. В інтересах ясності та ефективності та беручи до уваги обсяг і технічний характер змін до положень Регламенту (ЄС) № 273/2008, відповідні положення зазначеного Регламенту необхідно інкорпорувати в Імплементаційний регламент (ЄС) 2016/1240.
(3) Щоб забезпечити уніфіковане дотримання нових стандартів і методів у всіх державах-членах, лабораторіям необхідно надати достатній період часу для перегляду процедур та застосування оновлених методів.
(4) Тому необхідно внести відповідні зміни до Імплементаційного регламенту (ЄС) № 2016/1240.
(5) В інтересах правової визначеності Регламент (ЄС) № 273/2008 необхідно скасувати.
(6) Інструменти, передбачені в цьому Регламенті, відповідають висновку Керівного комітету з питань спільної організації сільськогосподарських ринків,
До Регламенту (ЄС) № 2016/1240 внести такі зміни:
(1) до статті 4 внести такі зміни:
(a) до параграфа 1 внести такі зміни:
(i) пункт (d) викласти у такій редакції:
«(d) для масла: у частинах I та Ia додатка IV до цього Регламенту»;
(ii) пункт (e) викласти у такій редакції:
«(e) для сухого знежиреного молока: у частинах I та Ia додатка V до цього Регламенту»;
(b) параграф 2 викласти у такій редакції:
«2. Методами, які мають використовуватися для визначення якості зернових культур, масла та сухого знежиреного молока, що підпадає під застосування державних інтервенцій, про які йдеться в додатках I, IV і V відповідно, повинні бути методи, що встановлені найостаннішими версіями відповідних європейських або міжнародних стандартів, у відповідних випадках, введених в дію щонайменше за 6 місяців до першого дня періоду державних інтервенцій, як визначено у статті 12 Регламенту (ЄС) № 1308/2013.»;
(2) текст доповнити статтею 60a такого змісту:
«Стаття 60a
Спеціальні положення щодо перевірок, пов’язаних із державними інтервенціями та допомогою на зберігання молока і молочних продуктів на складах приватного користування
1. Допустимість масла, сухого знежиреного молока та сиру для отримання допомоги на зберігання на складах приватного користування установлюється згідно з методами, визначеними у додатках VI, VII і VIII відповідно.
Такі методи встановлюються на основі найостанніших версій відповідних європейських або міжнародних стандартів, у відповідних випадках, введених в дію щонайменше за 6 місяців до першого дня періоду державних інтервенцій, як визначено у статті 12 Регламенту (ЄС) № 1308/2013.
2. Результати перевірок, проведених із застосуванням методів, визначених у цьому Регламенті, оцінюються згідно з додатком IX.»;
(3) до додатків внести зміни згідно з додатком до цього Регламенту.
Регламент (ЄС) № 273/2008 скасувати.
Цей Регламент набуває чинності на сьомий день після дня його публікації в Офіційному віснику Європейського Союзу.
Цей Регламент обов'язковий у повному обсязі та підлягає прямому застосуванню в усіх державах-членах.
Вчинено у Брюсселі 30 січня 2018 року.
__________
(-1) OB L 347, 20.12.2013, с. 549.
(-2) Делегований регламент Комісії (ЄС) № 2016/1238 від 18 травня 2016 року на доповнення Регламенту Європейського Парламенту і Ради (ЄС) № 1308/2013 стосовно державних інтервенцій та допомоги на зберігання на складах приватного користування (OВ L 206, 30.07.2016, с. 15).
(-3) Імплементаційний регламент Комісії (ЄС) № 2016/1240 від 18 травня 2016 року про встановлення правил застосування Регламенту Європейського Парламенту і Ради (ЄС) № 1308/2013 стосовно державних інтервенцій та допомоги на зберігання на складах приватного користування (OВ L 206, 30.07.2016, с. 71).
(-4) Регламент Комісії (ЄС) № 273/2008 від 5 березня 2008 року про встановлення детальних правил застосування Регламенту Ради (ЄС) № 1255/1999 стосовно методів дослідження та оцінювання якості молока і молочних продуктів (OB L 88, 29.03.2008, с. 1).
До додатків до Імплементаційного регламенту (ЄС) № 2016/1240 внести такі зміни:
(1) до додатка IV внести такі зміни:
(a) у пункті 2 частини I другий підпараграф викласти в такій редакції:
«Кожну пробу оцінюють індивідуально. Повторне відбирання проб або повторне оцінювання заборонено.»;
(b) текст доповнити частиною Ia такого змісту:
Методи дослідження несолоного масла у цілях здійснення державних інтервенцій
|
Параметр |
Метод |
|
ISO 17189 або ISO 3727, частина 3 |
|
|
Вода |
ISO 3727, частина 1 |
|
Суха знежирена речовина |
ISO 3727, частина 2 |
|
Кислотність жиру |
ISO 1740 |
|
Пероксидне число |
ISO 3976 |
|
Немолочний жир |
ISO 17678 |
|
Органолептичні характеристики |
ISO 22935, частини 2 і 3 та наведена нижче оціночна таблиця. |
|
Зовнішній вигляд |
Консистенція |
Запах і смак |
|||
|
Бали |
Коментарі |
Бали |
Коментарі |
Бали |
Коментарі |
|
5 |
Дуже добре
|
5 |
Дуже добре
|
5 |
Дуже добре
|
|
4 |
Добре (без явних дефектів) |
4 |
Добре (без явних дефектів) |
4 |
Добре (без явних дефектів) |
|
1, 2 або 3 |
З деяким дефектом |
1, 2 або 3 |
З деяким дефектом |
1, 2 або 3 |
З деяким дефектом» |
(2) текст додатка V доповнити частиною Ia такого змісту:
Методи дослідження сухого знежиреного молока в цілях застосування державних інтервенцій
|
Параметр |
Метод |
|
Білок |
ISO 8968, частина 1 |
|
Жир |
ISO 1736 |
|
Вода |
ISO 5537 |
|
Кислотність |
ISO 6091 |
|
Лактати |
ISO 8069 |
|
Тест на фосфатазу |
ISO 11816, частина 1 |
|
Індекс нерозчинності |
ISO 8156 |
|
ADPI |
|
|
Мікроорганізми |
ISO 4833, частина 1 |
|
Маслянка |
Доповнення I |
|
Доповнення II і III |
|
|
ISO 8069 або інспекційні перевірки на місці |
|
|
ISO 22935, частини 2 і 3 |
СУХЕ ЗНЕЖИРЕНЕ МОЛОКО: КІЛЬКІСНЕ ВИЗНАЧЕННЯ ФОСФАТИДИЛСЕРИНУ ТА ФОСФАТИДИЛЕТАНОЛАМІНУ
Метод описує процедуру кількісного визначення фосфатидилсерину (PS) та фосфатидилетаноламіну (РЕ) у сухому знежиреному молоці (СЗМ) і підходить для виявлення сухої речовини маслянки у СЗМ.
Вміст PS + РЕ: масова частка субстанції, визначена з використанням зазначеної тут процедури. Результат виражається у міліграмах фосфатидилетаноламін дипальмітойлу (PEDP) на 100 г порошку.
Екстрагування метанолом амінофосфоліпідів з відновленого сухого молока. Визначення PS та РЕ як похідних ο-фталдіальдегіду (ОРА) методом обернено-фазової ВЕРХ (ОФ ВЕРХ) та флуоресцентним детектуванням. Кількісне визначення вмісту PS та РЕ у дослідній пробі шляхом зіставлення зі стандартною пробою, що містить відому кількість PEDP.
Усі реагенти повинні мати визнану аналітичну якість. Вода повинна бути дистильованою або принаймні еквівалентного ступеня чистоти, якщо не зазначено інше.
4.1. Стандартний матеріал: PEDP, чистотою щонайменше 99 %
Примітка: Стандартний матеріал зберігають за температури - 18 °С.
4.2. Реагенти для підготування стандартної проби та дослідної проби
4.2.1. Метанол потрібної для ВЕРХ якості
4.2.2. Хлороформ потрібної для ВЕРХ якості
4.2.3. Триптамін-моногідрохлорид
4.3. Реагенти для дериватизації ο-фталдіальдегідом
4.3.1. Гідроксид натрію, водний розчин 12 М
4.3.2. Борна кислота, водний розчин 0,4 М, доведений до pH 10,0 за допомогою гідроксиду натрію (4.3.1)
4.4.1. Еелюенти повинні бути приготовлені з використанням реагентів потрібної для ВЕРХ якості.
4.4.2. Вода потрібної для ВЕРХ якості
4.4.3. Метанол з флуориметрично перевіреною чистотою
5.1. Аналітичні ваги, здатні зважувати з точністю до 1 мг, з дискретністю 0,1 мг
5.2. Лабораторні стакани ємністю 25 та 100 мл
5.3. Дозатори, здатні відмірювати 1 та 10 мл
5.5. Градуйовані дозатори, здатні відмірювати 0,2, 0,5 та 5 мл
5.6. Мірні колби ємністю 10, 50 та 100 мл
5.7. Шприци ємністю 20 та 100 мкл
5.9. Центрифуга, здатна досягати 27000 × g
5.10. Скляні пробірки ємністю близько 5 мл
5.11. Градуйований циліндр ємністю 25 мл
5.12. pH-метр з точністю вимірювання до 0,1 одиниць pH
5.13.1. Градієнтна насосна система, здатна працювати при швидкості потоку 1,0 мл/хв та тиску 200 бар
5.13.2. Автосемплер з можливістю дериватизації
5.13.3. Нагрівач колонки, здатний підтримувати температуру колонки на рівні 30 °С ± 1 °С
5.13.4. Флуоресцентний детектор, здатний працювати за довжини хвилі збудження 330 нм та довжини хвилі випромінювання 440 нм
5.13.5. Інтегратор або програмне забезпечення з можливістю вимірювання площі піків
5.13.6. Колонка LiChrospher® - 100 (250 × 4.6 мм) або еквівалентна колонка, заповнена октадецилсіланом (C 18), розмір частинок 5 мкм.
Відбирання проб здійснюють згідно зі Стандартом ISO 707.
7.1. Приготування розчину внутрішнього стандарту
7.1.1. Відмірюють 30,0 ± 0,1 мг триптамін-моногідрохлориду (4.2.3) у мірну колбу ємністю 100 мл (5.6.) і доповнюють до мітки метанолом (4.2.1)
7.1.2. Відмірюють дозатором (5.3.) 1 мл цього розчину у мірну колбу ємністю 10 мл (5.6.) і доповнюють до мітки метанолом (4.2.1), щоб отримати концентрацію триптаміну 0,15 мМ
7.2. Приготування розчину дослідної проби
7.2.1. Відмірюють 1,000 ± 0,001 г проби СЗМ у лабораторну склянку ємністю 25 мл (5.2.). Додають дозатором (5.3.) 10 мл дистильованої води температурою 40 °С ± 1 °С і змішують магнітним змішувачем (5.4.) протягом 30 хвилин, щоб розчинити всі грудки.
7.2.2. Відмірюють дозатором (5.5.) 0,2 мл відновленого молока у мірну колбу ємністю 10 мл (5.6.), за допомогою шприца (5.7.) додають 100 мкл розчину триптаміну 0,15 мМ (7.1.) і доповнюють до об’єму метанолом (4.2.1). Ретельно перемішують шляхом перевертання колби та обробляють ультразвуком (5.8.) протягом 15 хвилин.
7.2.3. Центрифугують (5.9.) за 27000 g × g протягом 10 хвилин і збирають надосадову рідину у скляну пробірку (5.10.)
Примітка: Розчин дослідної проби необхідно зберігати за температури 4 °С до проведення дослідження методом ВЕРХ.
7.3. Приготування розчину зовнішнього стандарту
7.3.1. Відмірюють 55,4 мг PEDP (4.1.) у мірну колбу ємністю 50 мл (5.6.) і додають приблизно 25 мл хлороформу (4.2.2) за допомогою градуйованого циліндра (5.11.). Нагрівають закорковану колбу до 50 °С ± 1 °С і ретельно перемішують, доки PEDP не розчиниться. Охолоджують колбу до 20 °С. доводять до об'єму метанолом (4.2.1) і перемішують шляхом перевертання.
7.3.2. Відмірюють дозатором (5.3.) 1 мл цього розчину у мірну колбу ємністю 100 мл (5.6.) і доводять до об'єму метанолом (4.2.1). Відмірюють дозатором (5.3.) 1 мл цього розчину у мірну колбу ємністю 10мл (5.6), додають 100 мкл (5.7.) розчину триптаміну 0,15 мМ (7.1.) і доводять до об'єму метанолом (4.2.1). Перемішують шляхом перевертання
Примітка: Розчин референтного зразка необхідно зберігати за температури 4 °С до проведення дослідження методом ВЕРХ.
7.4. Приготування дериватизаційного реагенту
Відмірюють 25,0 ±0,1 мг ОРА (4.3.4) у мірну колбу ємністю 10 мл (5.6.), додають дозатором (5.5.) 0.5 мл метанолу (4.2.1) і ретельно перемішують, щоб розчинити ОРА. Доповнюють до мітки розчином борної кислоти (4.3.2) і додають 20 мкл 2-меркаптоетанолу (4.3.3) за допомогою шприца (5.7.).
Примітка: Дериватизаційний реагент необхідно зберігати за температури 4 °С в ампулі з темного скла, так він залишається стабільним протягом одного тижня.
Розчинник A: Розчин дигідрофосфату натрію 0,3 мМ і розчин ацетату натрію 3 мМ (доведений до pH 6,5 ±0,1 за допомогою оцтової кислоти): метанол: тетрагідрофуран = 558:440:2 (об/об/об)
7.5.2. Пропонований градієнт елюювання:
|
Час (хв) |
Розчинник A (%) |
Розчинник B (%) |
Швидкість потоку (мл/хв) |
|
Початковий |
40 |
60 |
0 |
|
0,1 |
40 |
60 |
0,1 |
|
5,0 |
40 |
60 |
0,1 |
|
6,0 |
40 |
60 |
1,0 |
|
6,5 |
40 |
60 |
1,0 |
|
9,0 |
36 |
64 |
1,0 |
|
10,0 |
20 |
80 |
1,0 |
|
11,5 |
16 |
84 |
1,0 |
|
12,0 |
16 |
84 |
1,0 |
|
16,0 |
10 |
90 |
1,0 |
|
19,0 |
0 |
100 |
1,0 |
|
20,0 |
0 |
100 |
1,0 |
|
21,0 |
40 |
60 |
1,0 |
|
29,0 |
40 |
60 |
1,0 |
|
30,0 |
40 |
60 |
0 |
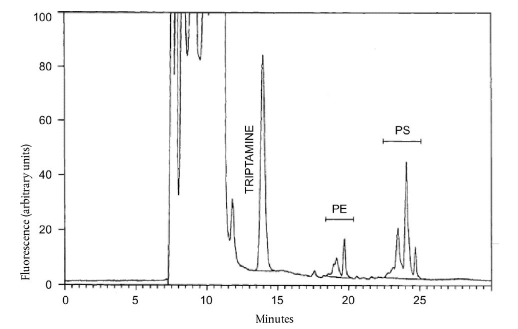
Примітка: Градієнт елюювання може потребувати незначної модифікації, щоб досягти розділення, показаного на рисунку 1.
7.5.3. Об’єм введення: 50 мкл дериватизаційногореагенту та 50 мкл розчину проби
Перед щоденним запуском системи колонку промивають 100 % розчином B протягом 15 хвилин, потім встановлюють співвідношення A:B = 40:60 і врівноважують при 1 мл/хв протягом 15 хвилин. Виконують холостий цикл з уведенням метанолу (4.2.1).
Примітка: Перед тривалим зберіганням колонку промивають розчином метанолу: хлороформ = 80:20 (об/об) протягом 30 хвилин.
7.5.5. Визначають вміст PS + РЕ у дослідній пробі
7.5.6. Виконують послідовність хроматографічних досліджень, дотримуючи постійний часовий інтервал між циклами, щоб отримати постійні часи утримування. Вводять розчин зовнішнього стандарту (7.3.) на кожні 5-10 розчинів дослідної проби для обчислення фактора відгуку.
Примітка: Колонку необхідно очищувати промиванням 100 % розчином B (7.5.1) протягом щонайменше 30 хвилин через кожні 20-25 циклів.
PEDP елююється у вигляді одиночного піка. Визначте площу піків шляхом їх інтегрування від западини до западини.
Триптамін елююється у вигляді одиночного піку (рисунок 1). Визначте площу піків шляхом інтегрування від западини до западини.
За описаних умов (рисунок 1), PS елююється у вигляді двох головних, частково нерозділених піків, яким передує менший пік. РЕ елююється у вигляді трьох головних, частково нерозділених піків. Визначають повну площу кожного кластеру піків, окреслюючи базову лінію, як представлено на рисунку 1.
8. ОБЧИСЛЕННЯ ТА ВИРАЖЕННЯ РЕЗУЛЬТАТІВ
Вміст PS + РЕ у дослідній пробі обчислюється таким чином:
C = 55,36 × ((А2)/(А1)) × ((Т1)/(Т2))
C = Вміст PS або РЕ (мг/100 г порошку) у дослідній пробі
А1 = площа піків PEDP розчину стандартної проби (7.3.)
А2 = площа піка PS або РЕ розчину дослідної проби (7.2.)
Т1 = Площа піків триптаміну розчину стандартної проби (7.3.)
Т2 = Площа піка триптаміну розчину дослідної проби (7.2.)
Примітка: Значення повторюваності були обчислені відповідно до Міжнародного стандарту IDF (*).
Відносний стандартний відхил повторюваності, що виражає варіативність незалежних аналітичних результатів, отриманих тим самим оператором, з використанням того самого обладнання за тих самих умов для дослідження тієї самої дослідної проби і в короткий інтервал часу повинен не перевищувати 2 % відносного значення. Якщо за цих умов отримані два визначення, відносна різниця між двома результатами повинна бути не більше ніж 6 % від середнього арифметичного результатів.
Якщо два визначення отримані операторами у різних лабораторіях з використанням різного обладнання, за різних умов для дослідження тієї самої дослідної проби, відносна різниця між двома результатами повинна бути не більше ніж 11 % від середнього арифметичного результатів.
10. ПЕРЕЛІК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ
10.1. Resmini Р., Pellegrino L., Hogenboom J.A., Sadini V., Rampilli M., ‘Detection of buttermilk solids in skimmilk powder by HPLC quantification of aminophospholipids’. Sci. Tecn. Latt.- Cas., 39,395 (1988).
Хроматограма ВЕРХ похідних ОРА фосфатидилсерину (PS) та фосфатидилетаноламіну (РЕ) у метаноловому екстракті відновленого сухого знежиреного молока. Представлений метод інтегрування для піків PS, РЕ та триптаміну (внутрішній стандарт)
ВИЯВЛЕННЯ СИЧУЖНОЇ СИРОВАТКИ У СУХОМУ ЗНЕЖИРЕНОМУ МОЛОЦІ ДЛЯ ДЕРЖАВНОГО ЗБЕРІГАННЯ, ШЛЯХОМ ВИЗНАЧЕННЯ КАЗЕЇНОМАКРОПЕПТИДІВ МЕТОДОМ ВИСОКОЕФЕКТИВНОЇ РІДИННОЇ ХРОМАТОГРАФІЇ (ВЕРХ)
1. СФЕРА ОХОПЛЕННЯ І ЗАСТОСУВАННЯ
Цей метод уможливлює виявлення сичужної сироватки у сухому знежиреному молоці, призначеному для державного зберігання, шляхом визначення казеїномакропептидів.
Міжнародний стандарт ISO 707 - Молоко та молочні продукти - Настанови щодо відбирання проб.
Вміст сухої речовини сичужної сироватки визначають як масову частку, визначену за вмістом казеїномакропептидів в рамках описаної процедури.
- Відновлення сухого знежиреного молока, видалення жиру та білків за допомогою трихлороцтової кислоти з наступним центрифугуванням або фільтруванням;
- Визначення кількості казеїномакропептидів (СМР) у надосадовій рідині методом високоефективної рідинної хроматографії (ВЕРХ);
- Оцінювання результатів дослідження проб на основі порівняння (звіряння) з результатами дослідження стандартних проб сухого знежиреного молока з доданням або без додання відомого відсотка сухої сироватки.
Усі реагенти повинні мати визнану аналітичну якість. Використовувана вода повинна бути дистильованою або принаймні еквівалентного ступеня чистоти.
5.1. Розчин трихлороцтової кислоти
Розчиняють 240 г трихлороцтової кислоти (CCl3COOH) у воді і доводять до 1000 мл. Розчин повинен бути прозорим і безбарвним.
Розчиняють 1,74 г гідроортофосфату калію (К2НРО4), 12,37 г дигідроортофосфату калію (КН2РО4) і 21,41 г сульфату натрію (Na2SO4) у приблизно 700 мл води. У разі необхідності, доводять до pH 6,0, використовуючи розчин фосфорної кислоти або гідроксиду калію.
Доводять до 1000 мл водою та гомогенізувати.
Примітка: Склад елюенту може бути оновлений, щоб відповідати сертифікату стандартів або рекомендаціям виробників матеріалів для наповнення колонок.
Перед використанням фільтрують елюент через мембранний фільтр з діаметром пор 0,45 мкм.
Змішують один об’єм ацетонітрилу (CH3CN) і дев’ять об’ємів води. Перед використанням фільтрують суміш через мембранний фільтр з діаметром пор 0,45 мкм.
Примітка: Може бути використаний будь-який інший розчин для промивання з бактерицидною дією, який не погіршує роздільну здатність колонок.
5.4.1. Сухе знежирене молоко, що відповідає вимогам цього Регламенту (тобто [0])
5.4.2. Таке саме сухе знежирене молоко з домішками 5 % (м/м) сичужного типу сухої сироватки стандартного складу (тобто [5])
6.2. Необов’язково: центрифуга, здатна досягати відцентрової сили 2200 g, оснащена центрифужними пробірками, закоркованими або з накінечниками, ємністю приблизно 50 мл
6.5. Скляні лійки, діаметром приблизно 7 см
6.6. Фільтрувальний папір, середньої фільтрації, діаметром приблизно 12,5 см
6.7. Скляне фільтрувальне обладнання з мембранним фільтром з діаметром пор 0,45 мкм
6.8. Градуйовані дозатори, що дають змогу відмірювати 10 мл (ISO 648, клас A, або ISO/R 835) або дозувальна система, здатна відмірювати 10,0 мл за дві хвилини
6.9. Дозувальна система, здатна відмірювати 20,0 мл води за температури приблизно 50 °С
6.10. Водяна баня-термостат, установлена на температуру 25 ± 0,5 °С
6.11. Обладнання для ВЕРХ, що складається з:
6.11.2. Інжектора, ручного або автоматичного, ємністю від 15 до 30 мкл
6.11.3. Двох з’єднаних послідовно колонок TSK 2000-SW (довжина 30 см, внутрішній діаметр 0,75 см) або еквівалентних колонок (напр. одинична колонка TSK 2000-SWxl, одинична колонка Agilent Technologies Zorbax GF 250) та передколонки (3 см ґ 0,3 см), заповнених I 125 або матеріалом з еквівалентною ефективністю
6.11.4. Термостатичної печі для колонки, установленої на температуру 35 ± 1 °С
6.11.5. УФ детектора зі змінною довжиною хвилі, можливістю вимірювання при 205 нм з чутливістю 0,008 А
6.11.6. Інтегратора з можливістю інтегрування від западини до западини
Примітка: Допускається робота з колонками, утримуваними за кімнатної температури, але їхня роздільна здатність є дещо нижчою. У такому випадку, коливання температури в рамках будь-якої однієї серії досліджень повинне бути менше ніж ± 5 °С.
7.1. Проби відбираються згідно з процедурою, встановленою у Міжнародному стандарті ISO 707. Проте, держави-члени можуть використовувати інший метод відбирання проб за умови, що він відповідає принципам вищезгаданого стандарту.
7.2. Пробу зберігають в умовах, які виключають будь-яке погіршення якості чи зміну складу.
8.1. Приготування розчину дослідної проби
Поміщають сухе молоко в контейнер, ємністю приблизно вдвічі більшою ніж об'єм сухої речовини, оснащений герметичною кришкою. Негайно закривають контейнер. Добре перемішують сухе молоко шляхом багаторазового перевертання контейнера.
Відмірюють 2,000 ± 0,001 г дослідної проби у центрифужну пробірку (6.2.) або відповідну колбу з корком (50 мл).
8.3.1 До аналітичної наважки додають 20,0 мл теплої води (50 °С). Розчиняють порошок шляхом струшування протягом п’яти хвилин з використанням механічного шейкера (6.3.). Пробірку поміщають у водяну баню (6.10.) і дають температурі вирівнятися до 25 °С
8.3.2. Додають поступово протягом двох хвилин 10,0 мл розчину трихлороцтової кислоти (5.1.) температурою приблизно 25 °С, одночасно енергійно перемішуючи за допомогою магнітної мішалки (6.4.). Пробірку поміщають у водяну баню (6.10.) і залишають на 60 хвилин.
8.3.3. Центрифугують (6.2.) за 2200 g протягом 10 хвилин, або фільтрують через фільтрувальний папір (6.6.), видаляючи перші 5 мл фільтрату
8.4. Хроматографічне визначення
8.4.1. Уводять від 15 до 30 мкл точно відміряної надосадової рідини або фільтрату (8.3.3) в рідинний хроматограф (6.11.), що працює за швидкості потоку 1,0 мл елюенту (5.2.) на хвилину
Примітка 1: Може бути застосована інша швидкість потоку, залежно від внутрішнього діаметру використовуваних колонок або інструкцій виробника колонки.
Примітка 2: Необхідно споліскувати колонки водою під час кожного переривання. Ніколи не можна залишати в них елюент (5.2.).
Перед будь-яким перериванням тривалістю більше ніж 24 години, споліскують колонки водою, потім промивають розчином (5.3.) протягом щонайменше трьох годин за швидкості потоку 0,2 мл на хвилину.
8.4.2. Результати хроматографічного дослідження дослідної проби [Е] отримують у вигляді хроматограми, на якій кожен пік ідентифікований за його часом утримування RT таким чином:
|
Пік II: |
Другий пік хроматограми з RT приблизно 12,5 хвилин. |
|
Пік III: |
Третій пік хроматограми, що відповідає СМР, з RT 15,5 хвилин. |
Вибір колонки (колонок) може мати істотний вплив на часи утримування окремих піків.
Інтегратор (6.11.6) автоматично обчислює площу А кожного піку:
Важливо уважно розглянути вигляд кожної хроматограми перед початком кількісної інтерпретації, щоб виявити будь-які аномалії, спричинені або несправністю обладнання чи колонок, або походженням чи природою досліджуваної проби.
Якщо виникають сумніви, дослідження повторюють.
8.5.1. Для стандартних проб (5.4.) точно дотримуються процедури, описаної в пунктах 8.2-8.4.2
Використовують щойно приготовані розчини, оскільки СМР розкладається у 8 % трихлороцтовому середовищі. Втрати оцінюються в 0,2 % на годину за 30 °С.
8.5.2. Перед початком хроматографічного дослідження проб, колонки кондиціонують шляхом багаторазового введення стандартної проби (5.4.2) у розчин (8.5.1), доки площа і час утримування піка, що відповідає СМР, не стануть постійними.
8.5.3. Визначають фактори відгуку R шляхом введення такого самого об’єму фільтратів (8.5.1), який використовувався для проб.
9.1. Метод обчислення та формули
9.1.1. Обчислення факторів відгуку R:
RII = фактори відгуку піків II,
AII [0] = площі піків II стандартної проби [0], отримані в 8.5.3.
АIII [0] and АIII [5] = площі піка III у стандартних пробах [0] і [5], отримані відповідно в 8.5.3,
W = кількість сироватки у стандартній пробі [5], тобто 5.
9.1.2. Обчислення відносної площі піків у пробі [E]
SII [E], SIII [E], SIV = відносні площі піків II, III і VI відповідно у пробі [E],
АII [Е], АIII [Е] = площі піків II і III відповідно у пробі [Е], отримані в 8.4.2,
RII, RIII = фактори відгуку, обчислені в 9.1.1.
9.1.3. Обчислення відносного часу утримування піка III у пробі [Е]:
RRTIII[E] = (RTIII[E])/(RTIII[5])
RRTIII[Е] = відносний час утримування піка III у пробі [Е],
RTIII[Е] = час утримування піка III у пробі [Е], отриманий у 8.4.2,
9.4.1. Експерименти показали, що є лінійна залежність між відносним часом утримування піка III, тобто RRTIII[Е], і відсотковим вмістом сухої сироватки, доданої до 10 %
- RRTIII[Е] є < 1,000, коли вміст сироватки є > 5 %;
- RRTIII[Е] є ≥ 1,000, коли вміст сироватки є ≤ 5 %.
Допустима невизначеність для значень RRTIII становить ± 0.002.
Зазвичай значення RRTIII[0] мало відхиляється від 1,034. Залежно від стану колонок, це значення може наближатись до величини 1,000, але завжди повинне її перевищувати.
9.2. Обчислення відсоткового вмісту сичужної сухої сироватки у пробі:
W = SIII[E] - [1,3 + (SIII[0] -0,9)]
|
W |
= відсотковий вміст м/м сичужної сироватки у пробі [Е]; |
|
SIII [Е] |
= відносна площа піка III дослідної проби [Е], отримана як у 9.1.2; |
|
1,3 |
= представляє відносну середню площу піка III, виражену в грамах сичужної сироватки на 100 г, визначену в сухому знежиреному молоці без домішок різного походження. Цю величину було отримано експериментальним шляхом; |
|
SIII [0] |
= представляє відносну площу піка III, яка дорівнює RIII × АIII [0]. Ці значення отримані у 9.1.1 та 8.5.3 відповідно; |
|
(SIII [0] -0,9) |
= представляє поправку, яку необхідно застосувати до відносної середньої площі 1,3, коли SIII [0] не дорівнює 0,9. Експериментальним шляхом визначено, що відносна середня площа піка III контрольної проби [0] становить 0,9. |
Різниця між результатами двох визначень, проведених одночасно або безпосередньо одне за одним тим самим аналітиком з використанням того самого обладнання на ідентичному дослідному матеріалі, повинна не перевищувати 0,2 % м/м.
Різниця між двома одиничними і незалежними результатами, отриманими у двох різних лабораторіях на ідентичному дослідному матеріалі, повинна не перевищувати 0,4 % м/м.
Припускають відсутність сироватки, якщо відносна площа піка III, SIII[Е], виражена в грамах сичужної сироватки на 100 г продукту, становить ≤ 2.0 + (SIII[0] - 0,9).
|
2,0 |
це максимальне допустиме значення для відносної площі піка III з урахуванням відносної середньої площі піка III, тобто 1,3, невизначеності через зміни складу сухого знежиреного молока та відтворюваності методу (9.3.2), |
|
(SIII[0] - 0,9) |
це поправка, яку необхідно застосувати, коли площа SIII [0] не дорівнює 0,9 (див. пункт 9.2.) |
9.4.2. Якщо відносна площа піка III, SIII [Е], є > 2.0 + (SIII[0] - 0.9), а відносна площа піка III, SII [Е] ≤ 160, вміст сичужної сироватки визначають як вказано в пункті 9.2.
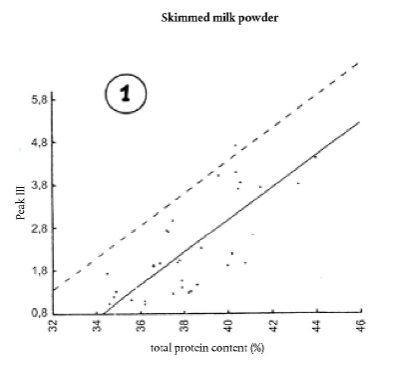
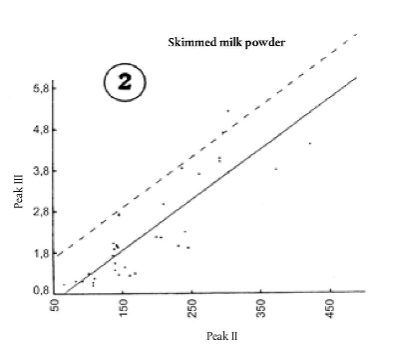
9.4.3. Якщо відносна площа піка III, SIII [Е], є > 2.0 + (SIII[0] - 0.9), а відносна площа піка II, IIn [Е] ≤ 160, визначають загальний вміст білка (Р %); потім розглядають графіки 1 і 2.
9.4.3.1. Дані, отримані після дослідження проб сухого знежиреного молока без домішок із високим загальним вмістом білка, згруповані на графіках 1 і 2.
Суцільна лінія представляє лінійну регресію, коефіцієнти якої обчислюються методом найменших квадратів.
Пунктирна пряма лінія визначає верхню межу відносної площі піка III з імовірністю того, що її не буде перевищено у 90 % випадків.
Пунктирні прямі лінії на графіках 1 і 2 мають такі рівняння:
SIII це відносна площа піка III, обчислена або за загальним вмістом білка, або за відносною площею піка SII [Е],
Р % це загальний вміст білка, виражений як відсотковий вміст, за масою,
SII [Е] це відносна площа проби, обчислена в пункті 9.1.2.
Ці рівняння еквівалентні величині 1,3, згаданій у пункті 9.2.
Розбіжність (Т1 та Т2) між установленою відносною площею SIII [Е] та відносною площею SIII виражається таким способом: Т1 = SIII[E] - [(0,376 Р % - 10,7) + (SIII[0] - 0,9)]Т2 = SIII[E] - (0,0123 SII[E] + 0,93) + (SII[0] - 0,9)]
|
Якщо Т1 та/або Т2 |
дорівнюють нулю або менше нуля, присутність сичужної сироватки не можна визначити. |
|
Якщо T1 та Т2 |
більше нуля, є присутність сичужної сироватки. |
Вміст сичужної сироватки обчислюється за такою формулою: W = Т2 + 0.91
0,91 - це відстань по вертикальній осі між суцільною і пунктирною прямими лініями.
ВИЗНАЧЕННЯ СУХОЇ РЕЧОВИНИ СИЧУЖНОЇ СИРОВАТКИ У СУХОМУ ЗНЕЖИРЕНОМУ МОЛОЦІ
1. МЕТА: ВИЯВЛЕННЯ ДОМІШОК СУХОЇ РЕЧОВИНИ СИЧУЖНОЇ СИРОВАТКИ ДО СУХОГО ЗНЕЖИРЕНОГО МОЛОКА
2. ПЕРЕЛІК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ: МІЖНАРОДНИЙ СТАНДАРТ ISO 707
Вміст сухої речовини сичужної сироватки визначають як масову частку, визначену за вмістом казеїномакропептидів в рамках описаної процедури.
Проби досліджують на казеїномакропептид A в рамках процедури обернено-фазової високоефективної рідинної хроматографії (процедура ВЕРХ). Оцінювання результатів дослідження проб на основі порівняння (звіряння) з результатами дослідження стандартних проб сухого знежиреного молока з доданням або без додання відомого відсотка сухої сироватки. Результати вище ніж 1 % (м/м) вказують на присутність сухої речовини сичужної сироватки.
Усі реагенти повинні мати визнану аналітичну якість. Використовувана вода повинна бути дистильованою або принаймні еквівалентного ступеня чистоти. Ацетонітрил повинен бути спектроскопічної якості або потрібної для ВЕРХ якості.
5.1. Розчин трихлороцтової кислоти
Розчиняють 240 г трихлороцтової кислоти (CCl3COOH) у воді і доводять до 1000 мл. Розчин повинен бути прозорим і безбарвним.
Елюент A: 150 мл ацетонітрилу (CH3CN), 20 мл ізопропанолу (СН3СНОНСН3) та 1,00 мл трифтороцтової кислоти (TFA, CF3COOH) поміщають у мірну колбу ємністю 1000 мл. Доводять до 1000 мл водою.
Елюент B: 550 мл ацетонітрилу, 20 мл ізопропанолу та 1,00 мл TFA поміщають у мірну колбу ємністю 1000 мл. Доводять до 1000 мл водою. Перед використанням фільтрують елюент через мембранний фільтр з діаметром пор 0,45 мкм.
Після проведення досліджень колонку промивають елюентом B (за допомогою градієнта), а потім промивають ацетонітрилом (за допомогою градієнта протягом 30 хвилин). Колонку зберігають в ацето нітрилі.
5.4.1. Сухе знежирене молоко, що відповідає вимогам для державного зберігання (тобто [0]).
5.4.2. Таке саме сухе знежирене молоко з домішками 5 % (м/м) сичужного типу сухої сироватки стандартного складу (тобто [5]).
5.4.3. Таке саме сухе знежирене молоко з домішками 50 % (м/м) сичужного типу сухої сироватки стандартного складу (тобто [50]).
6.2. Необов’язково: центрифуга, здатна досягати відцентрової сили 2200 g, оснащена центрифужними пробірками, закоркованими або з накінечниками, ємністю приблизно 50 мл
6.5. Скляні лійки, діаметром приблизно 7 см
6.6. Фільтрувальний папір, середньої фільтрації, діаметром приблизно 12,5 см
6.7. Скляне фільтрувальне обладнання з мембранним фільтром з діаметром пор 0,45 мкм
6.8. Градуйовані дозатори, що дають змогу відмірювати 10 мл (ISO 648, клас A, або ISO/R 835), або дозувальна система, здатна відмірювати 10,0 мл за дві хвилини
6.9. Дозувальна система, здатна відмірювати 20,0 мл води за температури приблизно 50 °С
6.10. Водяна баня-термостат, установлена на температуру 25 ± 0,5 °С
6.11. Обладнання для ВЕРХ, що складається з:
6.11.1. Бінарної градієнтної насосної системи
6.11.2. Інжектора, ручного або автоматичного, ємністю 100 мкл
6.11.3. Колонки Agilent Technologies Zorbax 300 SB-C3 (довжина 25 см, внутрішній діаметр 0,46 см) або еквівалентної колонки на базі широкопористого силікагелю для обернено-фазової хроматографії
6.11.4. Термостатичної печі для колонки, установленої на температуру 35 ± 1 °С
6.11.5. УФ детектора зі змінною довжиною хвилі, можливістю вимірювання при 210 нм (якщо необхідно, можна використовувати вище значення довжини хвилі до 220 нм) з чутливістю 0,02 А
6.11.6. Інтегратора з можливістю інтегрування по спільній базовій лінії або від западини до западини
Примітка: Допускається робота колонки за кімнатної температури за умови, що кімнатна температура не коливається більш ніж на 1 °С, інакше спостерігається занадто велика варіація в часі утримування СМРА.
7.1. Проби відбираються згідно з процедурою, встановленою у Міжнародному стандарті ISO 707. Проте, держави-члени можуть використовувати інший метод відбирання проб за умови, що він відповідає принципам вищезгаданого стандарту.
7.2. Пробу зберігають в умовах, які виключають будь-яке погіршення якості чи зміну складу.
8.1. Приготування розчину дослідної проби
Поміщають сухе молоко в контейнер, ємністю приблизно вдвічі більшою ніж об'єм сухої речовини, оснащений герметичною кришкою. Негайно закривають контейнер. Добре перемішують сухе молоко шляхом багаторазового перевертання контейнера.
Відмірюють 2,00 ± 0,001 г дослідної проби у центрифужну пробірку (6.2.) або відповідну колбу з пробкою (50 мл).
Примітка: У випадку сумішей, відмірюють таку кількість дослідної проби, щоб знежирена наважка проби відповідала 2,00 г.
8.3.1. До аналітичної наважки додають 20,0 мл теплої води (50°С). Розчиняють порошок шляхом струшування протягом п’яти хвилин з використанням механічного шейкера (6.3.). Пробірку поміщають у водяну баню (6.10.) і дають температурі вирівнятися до 25°С
8.3.2. Додають поступово протягом двох хвилин 10,0 мл розчину трихлороцтової кислоти (5.1.) температурою приблизно 25 °С, одночасно енергійно перемішуючи за допомогою магнітної мішалки (6.4.). Пробірку поміщають у водяну баню (6.10.) і залишають на 60 хвилин.
8.3.3 Центрифугують (6.2.) за 2200 g протягом 10 хвилин, або фільтрують через фільтрувальний папір (6.6.), видаляючи перші 5 мл фільтрату
8.4. Хроматографічне визначення
8.4.1. Метод обернено-фазової ВЕРХ виключає можливість отримання хибно-позитивних результатів завдяки присутності кислої маслянки в порошку.
8.4.2. Перед проведенням дослідження методом обернено-фазової ВЕРХ необхідно оптимізувати умови градієнта. Час утримування 26 ± 2 хвилини для СМРА є оптимальним для градієнтних систем з мертвим об'ємом близько 6 мл (об'єм від точки, в якій розчинники змішуються, до об'єму в петлі інжектора включно). Для градієнтних систем з меншим мертвим об'ємом (напр., 2 мл) необхідно застосовувати 22 хвилини як оптимальний час утримування.
Беруть розчини стандартних проб (5.4.) з доданням 50 % сичужної сироватки та без такого додання.
Уводять 100 мкл надосадової рідини або фільтрату (8.3.3) в рідинних хроматограф, що працює за умов градієнта, виявленого емпіричним шляхом, наведених у таблиці 1.
Умови градієнта для оптимізації хроматографії
|
Час (хв) |
Потік (мл/хв) |
% A |
% B |
Крива |
|
Початковий |
1,0 |
90 |
10 |
* |
|
27 |
1,0 |
60 |
40 |
лінійна |
|
32 |
1,0 |
10 |
90 |
лінійна |
|
37 |
1,0 |
10 |
90 |
лінійна |
|
42 |
1,0 |
90 |
10 |
лінійна |
Порівняння двох хроматограм повинне виявити місце розташування піка СМРА.
Використовуючи наведену нижче формулу, початковий склад розчину, який має бути використаний для нормального градієнта (див. 8.4.3), можна обчислити як % B = 10 - 2,5 + (13,5 + (RTcmpA - 26)/6) * 30/27 % B = 7,5 + (13,5 + (RTcmpA- 26)/6) * 1,11
|
RTcmpA |
: |
час утримування СМРА у градієнті, виявленому емпіричним шляхом |
|
10 |
: |
початковий % B градієнта, виявленого емпіричним шляхом |
|
2,5 |
: |
% B у середній точці мінус % B на початку нормального градієнта |
|
13,5 |
: |
час у середній точці градієнта, виявленого емпіричним шляхом |
|
26 |
: |
потрібний час утримування СМРА |
|
6 |
: |
співвідношення нахилів градієнта, виявленого емпіричним шляхом, та нормального градієнта |
|
30 |
: |
% B на початку мінус % B на 27 хвилині градієнта, виявленого емпіричним шляхом |
|
27 |
: |
час перебігу градієнта, виявленого емпіричним шляхом |
8.4.3. Уведення розчинів дослідних проб
Уводять 100 мкл точно відміряної надосадової рідини або фільтрату (8.3.3) в рідинний хроматограф, що працює за швидкості потоку 1,0 мл елюенту (5.2.) на хвилину.
Склад елюенту на початку дослідження отримують з пункту 8.4.2. Зазвичай він близький до A:B = 76:24 (5.2.). Відразу після введення починається лінійний градієнт, який дає 5 % збільшення відсотка B через 27 хвилин. Потім починається лінійний градієнт, який доводить склад елюенту до 90 % B за п'ять хвилин. Цей склад утримується протягом п’яти хвилин, після чого, через лінійний градієнт, змінюється за п'ять хвилин до початкового складу. Залежно від внутрішнього об'єму насосної системи, наступне введення можна здійснювати через 15 хвилин після досягнення початкових умов.
Примітка 1: Час утримування СМРA повинен становити 26 ± 2 хвилини. Цього можна досягнути шляхом варіювання початкових та кінцевих умов першого градієнта. Проте, різниця у % B для початкових та кінцевих умов першого градієнта повинна залишатись на рівні 5 % B.
Примітка 2: Елюенти повинні бути дегазовані достатньою мірою і залишатись у такому стані. Це є суттєво важливим для належного функціонування градієнтної насосної системи. Стандартний відхил для часу утримування піка СМРA повинен бути меншим ніж 0,1 хвилини (n = 10).
Примітка 3: Через кожні п'ять проб необхідно вводити референтний зразок [5] і використовувати для обчислення нового фактора відгуку R. (9.1.1).
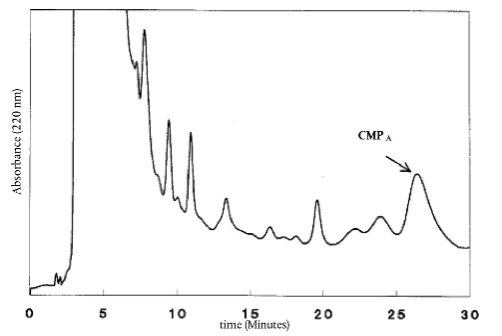
8.4.4. Результати хроматографічного дослідження дослідної проби (Е) отримують у вигляді хроматограми, на якій пік СМРА ідентифікують за його часом утримування у близько 26 хвилин.
Інтегратор (6.11.6) автоматично обчислює висоту Н піка СМРA. Місце розташування базової лінії необхідно перевіряти у кожній хроматограмі. Дослідження або інтегрування необхідно повторити, якщо базова лінія неправильно розташована.
Примітка: Якщо пік СМРА достатньо відділений від інших піків, необхідно застосовувати базову лінію від западини до западини, в іншому випадку застосовують опущення перпендикулярів до спільної базової лінії, початкова точка яких повинна бути близько до піка СМРA (отже, не за t = 0 хв!). Використовують такий самий тип інтегрування для стандарту і для проб та, у випадку застосування спільної базової лінії, перевіряють її послідовність для проб та стандарту.
Важливо уважно розглянути вигляд кожної хроматограми перед початком кількісної інтерпретації, щоб виявити будь-які аномалії, спричинені або несправністю обладнання чи колонок, або походженням чи природою досліджуваної проби. Якщо виникають сумніви, дослідження повторюють.
8.5.1. Для стандартних проб (5.4.1-5.4.2) точно дотримуватися процедури, описаної в пунктах 8.2.-8.4.4. Використовують щойно приготовані розчини, оскільки СМР розкладається у 8 % трихлороцтовому середовищі за кімнатної температури. За 4 °С розчин залишається стабільним впродовж 24 годин. У випадку тривалих серій досліджень, бажано використовувати охолоджений лоток для проб в автоматичному інжекторі.
Примітка: Пункт 8.4.2. можна пропустити, якщо значення % В за початкових умов відоме з попередніх досліджень.
Хроматограма референтного зразка [5] повинна бути аналогічною до представленої на рисунку. 1. На цьому рисунку пікові СМРА передують два менші піки. Важливо отримати подібне розділення.
8.5.2. Перед початком хроматографічного визначення проб вводять 100 мкл стандартної проби без сичужної сироватки [0] (5.4.1)
Хроматограма не повинна показувати піка в час утримування СМРА
8.5.3. Визначають фактори відгуку R шляхом введення такого самого об’єму фільтрату (8.5.1), який використовувався для проб.
9.1. Метод обчислення та формули
9.1.1. Обчислення фактора відгуку R:
W = кількість сироватки у стандартній пробі [5].
9.2. Обчислення відсоткового вмісту сухої сичужної сироватки у пробі:
W(E) = відсотковий вміст (м/м) сичужної сироватки у пробі (Е).
R = фактор відгуку піка СМРА (9.1.1)
Н(Е) = висота піка СМРА проби (Е)
Якщо W(E) більше ніж 1 %, а різниця між часом утримування дослідної проби та часом утримування стандартної проби [5] є менша ніж 0,2 хвилини, це свідчить про присутність сухої речовини сичужної сироватки.
Різниця між результатами двох визначень, проведених одночасно або безпосередньо одне за одним тим самим аналітиком з використанням того самого обладнання на ідентичному дослідному матеріалі, повинна не перевищувати 0,2 % м/м.
В діапазоні від 0 до 16 % сичужної сироватки повинна бути отримана лінійна залежність з коефіцієнтом кореляції > 0,99.
Межа, що становить 1 %, включає невизначеність, пов’язану з відтворюваністю.
(*) Міжнародний IDF стандарт 135B/1991. Молоко та молочні продукти. Прецизійні характеристики аналітичних методів. Схема процедури спільного дослідження.»
(3) текст доповнити такими додатками:
Методи дослідження масла, що зберігається на складах приватного користування
|
Параметр |
Метод |
|
ISO 17189 або ISO 3727, частина 3 |
|
|
Вода |
ISO 3727, частина 1 |
|
Суха знежирена речовина (за винятком солі) |
ISO 3727, частина 2 |
|
Сіль |
ISO 15648 |
Методи дослідження сухого знежиреного молока, що зберігається на складах приватного користування
Методи дослідження сирів, що зберігаються на складах приватного користування
1. Метод дослідження, наведений у доповненні, використовується, щоб гарантувати, що сир, виготовлений виключно з овечого молока, козячого молока чи буйволячого молока , або з суміші овечого, козячого та буйволячого молока , не містить казеїну коров’ячого молока.
Вважається, що казеїн коров’ячого молока є присутнім у продукті, якщо вміст казеїну коров’ячого молока в досліджуваній пробі дорівнює вмісту чи перевищує його у референтному зразку, що містить 1 % коров’ячого молока, як зазначено у доповненні.
2. Методи виявлення казеїну коров’ячого молока у сирах, про які йдеться в параграфі 1, можуть бути використані за умови, що:
(a) межа детектування становить максимум 0,5 %, та
(b) немає хибно-позитивних результатів, та
(c) казеїн коров’ячого молока може бути виявлений з потрібною чутливістю, навіть після тривалих періодів дозрівання, що може відбуватися у звичайних комерційних умовах.
Якщо будь-яку зі згаданих вище вимог не дотримано, використовуються методи, наведені у доповненні.
МЕТОД ВИЯВЛЕННЯ КОРОВ’ЯЧОГО МОЛОКА ТА КАЗЕЇНАТУ У СИРАХ З ОВЕЧОГО МОЛОКА, КОЗЯЧОГО МОЛОКА ЧИ БУЙВОЛЯЧОГО МОЛОКА, АБО СУМІШЕЙ ОВЕЧОГО, КОЗЯЧОГО ТА БУЙВОЛЯЧОГО МОЛОКА
Виявлення коров’ячого молока та казеїнату у сирах, виготовлених з овечого молока, козячого молока, буйволячого молока або сумішей овечого, козячого та буйволячого молока методом ізоелектричного фокусування γ-казеїнів після плазмінолізу.
Метод придатний для чутливого та специфічного виявлення натурального і термічно обробленого коров’ячого молока та казеїнату у свіжих і дозрілих сирах, виготовлених з овечого молока, козячого молока, буйволячого молока або сумішей овечого, козячого та буйволячого молока. Він не придатний для виявлення підроблення молока та сиру концентратами термічно оброблених сироваткових білків коров’ячого молока.
3.1. Виділення казеїнів з сиру та контрольних стандартів
3.2. Розчинення виділених казеїнів та піддання їх розщепленню плазміном (ЕС.3.4.21.7)
3.3. Ізоелектричне фокусування оброблених плазміном казеїнів у присутності сечовини та забарвлювання білків
3.4. Оцінювання забарвлених смуг γ3-казеїну та γ2-казеїну (доказ наявності коров’ячого молока) шляхом порівняння смуги, отриманої з дослідної проби, зі смугами, отриманими у тому самому гелі з контрольних стандартів, що містять 0 % та 1 % коров’ячого молока.
Якщо не вказано інше, використовуються хімічні речовини аналітичної якості. Вода повинна бути подвійної дистиляції або еквівалентного ступеня чистоти.
Примітка: Наведені нижче дані стосуються лабораторно приготованих поліакриламідних гелів, що містять сечовину, з параметрами 265 × 125 × 0,25 мм. У випадку використання інших параметрів та типів гелю, може виникнути необхідність коригування умов розділення.
4.1. Реагенти для вироблення поліакриламідних гелів, що містять сечовину
0,15 г N, N’-метилен-біс-акриламіду (BIS)
у воді та доводять до 100 мл і зберігають в ампулі з темного скла в холодильнику.
Примітка: Доступний у продажу готовий акриламід/BIS розчин може бути використаний замість вказаних встановлених масових часток нейротоксичних акриламідів. Якщо такий розчин містить 30 % м/об акриламіду та 0,8 % м/об BIS, для приготування використовується об’єм у 16,2 мл. замість встановлених масових часток. Термін зберігання стокового розчину становить максимум 10 днів; якщо його провідність є вище ніж 5 мкСм, розчин деіонізують шляхом перемішування з 2 г Амберліту MB-3 протягом 30 хвилин, потім пропускають через мембранний фільтр з розміром пор 0,45 мкм.
Приготовляють розчин гелю шляхом змішування добавок та амфолітів (*) із стоковим розчином гелю (див. 4.1.1).
Розчин гелю перемішують і дегазують протягом двох-трьох хвилин в ультразвуковій ванні або у вакуумі.
Примітка: Приготовляють розчин гелю безпосередньо перед його заливанням (див. 6.2.).
4.1.3.1. N, N, N’ N’ - тетраметилетилендіамін (Temed)
4.1.3.2. 40 % м/об амоній персульфат (PER):
Розчиняють 800 мг PER у воді та доводять до 2 мл.
Примітка: Завжди використовують щойно приготований розчин PER.
Розчиняють 5,77 г фосфорної кислоти (85 % м/м) у воді та розводять до 100 мл.
Розчиняють 2,00 г гідроксиду натрію у воді та розводять водою до 100 мл.
4.5. Реагенти для виділення білків
4.5.1. Розведена оцтова кислота (25,0 мл льодяної оцтової кислоти, доведеної водою до 100 мл)
4.6. Буфер для розчинення білків
Примітка: Зберігають у холодильнику, максимальний термін зберігання - один тиждень.
4.7. Реагенти для розщеплення казеїнів плазміном
Титрують 0,2 моль/л розчин гідрогенкарбонату амонію (1,58 г/100 мл води), що містить 0,05 моль/л етилендіамінтетраоцтову кислоту (EDTA, 1,46г/100 мл), 0,2 моль/л розчином карбонату амонію (1,92 г/100 мл води), що містить 0,05 моль/л EDTA, до досягнення pH 8.
4.7.2. Бичачий плазмін (ЕС. 3.4.21.7), з активністю щонайменше 5 Од/мл
4.7.3. Розчин ε-амінокапронової кислоти для інгібування ферментів
Розчиняють 2,624 г ε-амінокапронової кислоти (6-аміно-n-гексанової кислоти) у 100 мл 40 % (об/ об)) етанолу.
4.8.1. Сертифіковані контрольні стандарти суміші сичужного овечого та козячого знежиреного молока, що містить 0 % і 1 % коров’ячого молока, можна отримати в Інституті еталонних матеріалів та вимірювань Комісії, В-2440 Геель, Бельгія
4.8.2. Підготування внутрішньолабораторних стандартів буйволячого молока з доданням сичужного ферменту , що містять 0 % і 1 % коров’ячого молока
Знежирене молоко приготовляють центрифугуванням або буйволячого або коров’ячого сирого молока за температури 37 °С при 2500 g протягом 20 хвилин. Після швидкого охолодження пробірки та її вмісту до 6-8 °С, верхній шар жиру видаляють повністю. Для підготування 1 % стандарту додають 5,00 мл коров’ячого знежиреного молока до 495 мл буйволячого знежиреного молока в 1-літровій лабораторній склянці, доводять до pH 6,4 доданням розведеної молочної кислоти (10 % м/об). Доводять температуру до 35 °С і додають 100 мкл телячого сичужного ферменту (активність ферменту 1:10000, близько 3000 Од/мл), мішають протягом 1 хвилини і потім залишають накриту алюмінієвою фольгою склянку на одну годину за температури 35 °С, щоб утворився згусток. Після утворення згустку, цільне молоко з доданням сичужного ферменту ліофілізують без попередньої гомогенізації чи зціджування сироватки. Після ліофілізації його тонко подрібнюють до отримання однорідного порошку. Для підготування 0 % стандарту, здійснюють таку саму процедуру з використанням натурального буйволячого знежиреного молока. Стандарти зберігають за температури - 20 °С.
Примітка: Рекомендується перевіряти чистоту буйволячого молока ізоелектричним фокусуванням оброблених плазміном казеїнів перед підготуванням стандартів.
Реагенти для забарвлювання білків
Розчиняють 150 г трихлороцтової кислоти у воді і доводять до 1000 мл.
Розводять 500 мл метанолу та 200 мл льодяної оцтової кислоти до 2000 мл дистильованою водою.
Примітка: Готують свіжий знебарвлювальний розчин кожного дня; його можна приготувати змішуванням рівних об’ємів стокових розчинів 50 % (об/об) метанолу та 20 % (об/об) льодяної оцтової кислоти.
4.11.1. Забарвлювальний розчин (стоковий розчин 1)
Розчиняють 3,0 г кумасі діамантового синього G-250 (C.I. 42655) в 1000 мл 90 % (об/об) метанолу з використанням магнітної мішалки (приблизно 45 хвилин), пропускають через два складені фільтри з середньою швидкістю фільтрації.
4.11.2. Забарвлювальний розчин (стоковий розчин 2)
Розчиняють 5,0 г пентагідрату сульфату міді в 1000 мл 20 % (об/об) оцтової кислоти.
4.11.3. Забарвлювальний розчин (робочий розчин)
Змішують разом по 125 мл кожного зі стокових розчинів (4.11.1, 4.11.2) безпосередньо перед забарвлюванням.
Примітка: Забарвлювальний розчин повинен бути приготований у той день, коли його мають використовувати.
5.1. Скляні пластини (265 × 125 × 4 мм); гумовий валик (ширина 15 см); вирівнювальний столик
5.2. Лист із нанесеним гелем (265 × 125 мм)
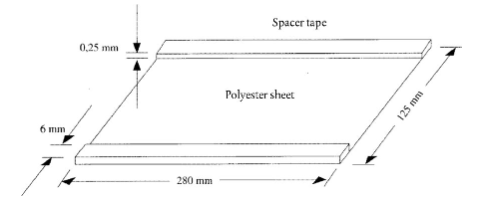
5.3. Покривний лист (280 × 125 мм). Приклеюють смужку клейкої стрічки (280 × 6 × 0,25 мм) до кожного довгого краю (див. рисунок 1)
5.4. Камера для електрофокусування з охоложувальною пластиною (напр., 265 × 125 мм) та відповідним джерелом живлення (≥ 2,5 кВ) або автоматичний електрофоретичний прилад
5.5. Циркуляційний кріостат, термостатично контрольований на 12 ± 0,5 °С
5.6. Центрифуга з регулюванням до 3000 g
5.7. Електродні смужки (довжиною ≥ 265 мм)
5.8. Пластикові пляшки-капанки для анодного та катодного розчинів
5.9. Аплікатори проб (10 × 5 мм, фільтрувальний папір, віскозний або з низькою адсорбцією білків)
5.10. Посуд з неіржавної сталі або зі скла для забарвлювання та знебарвлювання (напр., лотки для інструментів 280 × 150 мм)
5.12. Регульований гомогенізатор (діаметр стрижня 10 мм), з діапазоном від 8000 до 20000 об/хв
5.15. Апарат для зварювання плівки
5.16. Мікродозатори ємністю 25 мкл
5.17. Вакуум-концентратор або ліофілізатор
5.18. Водяна баня-термостат з регулятором, регульована до 35 та 40 ± 1 °С, з шейкером
5.19. Денситометричне обладнання з вимірюванням при λ = 634 нм
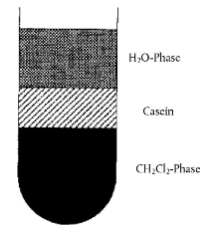
Відмірюють кількість, еквівалентну 5 грамам сухої маси сиру або контрольним стандартам, у 100 мл центрифужну пробірку, додають 60 мл дистильованої води і гомогенізують за допомогою стрижневого гомогенізатора (від 8000 до 10000 об/хв). Доводять до pH 4,6 розведеною оцтовою кислотою (4.5.1) та центрифугують (5 хвилин, 3000 g). Зливають жир і сироватку, гомогенізують залишок за 20000 об/хв у 40 мл дистильованої води, доведеної до pH 4,5 розведеною оцтовою кислотою (4.5.1), додають 20 мл дихлорметану (4.5.2), знову гомогенізують та центрифугують (5 хвилин, 3000 g). Шар казеїну між водною та органічною фазами (див. рисунок 2) видаляють шпателем і зливають обидві фази. Повторно гомогенізують казеїн у 40 мл дистильованої води (див. вище) і 20 мл дихлорметану (4.5.2) та центрифугують. Процедуру повторюють допоки обидві екстраговані фази не стануть безбарвними (два-три рази). Гомогенізують залишок білків з 50 мл ацетону (4.5.3) і пропускають через складений фільтрувальний папір з середньою швидкістю фільтрації. Змивають залишок на фільтрі двома окремими 25 мл порціями ацетону кожного разу і дають висохнути на повітрі або в струмені азоту, потім тонко подрібнюють у ступці.
Примітка: Сухі ізоляти казеїну необхідно зберігати за температури -20 °С.
6.1.2. Розщеплення плазміном β-казеїнів для інтенсифікації γ-казеїнів
Диспергують 25 мг виділених казеїнів (6.1.1) у 0,5 мл буферного розчину карбонату амонію (4.7.1) та гомогенізують протягом 20 хвилин, використовуючи, наприклад, оброблення ультразвуком. Підігрівають до 40 °С і додають 10 мкл плазміну (4.7.2), перемішують та інкубують протягом однієї години за температури 40 °С, постійно струшуючи. Щоб інгібувати фермент, додають 20 мкл розчину ε-амінокапронової кислоти (4.7.3), потім додають 200 мг сечовини у твердому стані та 2 мг дитіотреітолу.
Примітка: Щоб отримати кращу симетрію у фокусованих смугах казеїну, рекомендується ліофілізувати розчин після додання ε-амінокапронової кислоти і потім розчинити отримані ліофілізати у 0,5 мл буферу для розчинення білків (4.6.).
6.2. Приготування поліакриламідних гелів, що містять сечовину
Додавши декілька крапель води, розкочують валиком лист із нанесеним гелем (5.2.) на скляній пластині (5.1.), видаляючи надлишок води паперовим рушником або серветкою. Таким самим чином розкочують валиком покривний лист (5.3.) з прокладками (0,25 мм) на іншій скляній пластині. Кладуть пластину горизонтально на вирівнювальний столик.
Додають 10 мкл Temed (4.1.3.1) до підготовленого і деаерованого розчину гелю (4.1.2), перемішують і додають 10 мкл розчину PER (4.1.3.2), ретельно перемішують і відразу рівномірно виливають у центр покривного листа. Ставлять один край пластини з нанесеним гелем (нанесеною стороною донизу) на пластину з покривним листом і повільно опускають її таким чином, щоб між листами сформувалась і рівномірно розподілилась гелева плівка без бульбашок (рисунок 3). Обережно опускають пластину з нанесеним гелем повністю за допомогою тонкого шпателя і кладуть на неї зверху ще три скляні пластини, які діятимуть як гніт. Після завершення полімеризації (близько 60 хвилин) відділяють гель, що полімеризувався на листі із нанесеним гелем, разом з покривним листом, легко постукуючи по скляних пластинах. Обережно очищують зворотну сторону листа носія, щоб видалити залишки гелю та сечовини. Зварюють повздовжні краї «гелевого сендвіча», щоб отримати плівкову трубку, і зберігають у холодильнику (максимум шість тижнів).
Примітка: Покривний лист з прокладками може бути використаний повторно. Поліакриламідний гель можна порізати на пластини меншого розміру, що рекомендується, коли є кілька проб або використовується автоматичний електрофоретичний прилад (два гелі, розміром 4,5 × 5 см).
6.3. Ізоелектричне фокусування
Встановлюють охолоджувальний термостат на 12 °С. Протирають зворотну сторону листа із нанесеним гелем керосином, потім капають кілька крапель керосину (4.2.) у центр охолоджувального блока. Потім на ньому розкочують валиком «гелевий сендвіч», стороною листа носія донизу, не допускаючи утворення бульбашок. Витирають надлишок керосину і знімають покривний лист. Змочують електродні смужки електродними розчинами (4.3., 4.4.), обрізають по довжині гелю і поміщають у визначені місця (відстань між електродами 9,5 см).
Умови ізоелектричного фокусування:
6.3.1. Розмір гелю 265 × 125 × 0,25 мм
|
Етап |
Час (хв) |
Напруга (B) |
Сила струму (мA) |
Потужність (Вт) |
Вольт-години (В•год) |
|
1 .Попереднє фокусування |
30 |
максимум 2 500 |
максимум 15 |
постійна 4 |
прибл. 300 |
|
60 |
максимум 2 500 |
максимум 15 |
постійна 4 |
прибл. 1 000 |
|
|
3.Остаточне фокусування |
60 |
максимум 2 500 |
максимум 5 |
максимум 20 |
прибл. 3 000 |
|
|
40 |
максимум 2 500 |
максимум 6 |
максимум 20 |
прибл. 3 000 |
|
|
30 |
максимум 2 500 |
максимум 7 |
максимум 25 |
прибл. 3 000 |
Примітка: Якщо товщина чи ширина гелів змінюються, величини сили струму та потужності повинні бути відповідно скореговані (наприклад, подвоюють величини сили струму та потужності, якщо використовується гель розміром 265 × 125 × 0,5 мм).
6.3.2. Приклад програмування напруги для автоматичного електрофоретичного приладу (2 гелі розміром 5,0 × 4,5 см), електроди без смужок, прикладені безпосередньо до гелю
|
Етап |
Напруга |
Сила струму |
Потужність |
Температура |
Вольт-години |
|
1. Попереднє фокусування |
1 000 В |
10,0 мА |
3,5 Вт |
8 °С |
85 В•год |
|
2. Фокусування проби |
250 В |
5,0 мА |
2,5 Вт |
8 °С |
30 В•год |
|
3. Фокусування |
1 200 В |
10,0 мА |
3,5 Вт |
8 °С |
80 В•год |
|
4. Фокусування |
1 500 В |
5,0 мА |
7,0 Вт |
8 °С |
570 В•год |
Кладуть аплікатор проб на етапі 2 при 0 В•год.
Знімають аплікатор проб на етапі 2 при 30 В•год.
Виймають електродні смужки відразу після вимкнення живлення і негайно поміщають гель у посудину для забарвлювання/знебарвлювання, наповнену 200 мл фіксатора (4.9.); залишають на 15 хвилин, постійно струшуючи.
6.4.2. Промивання та забарвлювання пластини гелю
Старанно зціджують фіксатор і промивають пластину гелю два рази по 30 секунд у 100 мл знебарвлювального розчину (4.10.). Зливають знебарвлювальний розчин і наповнюють посудину 250 мл забарвлювального розчину (4.11.3); витримують протягом 45 хвилин, легко струшуючи.
6.4.3. Знебарвлювання пластини гелю
Зливають забарвлювальний розчин, промивають пластину гелю двічі, щоразу використовуючи 100 мл знебарвлювального розчину (4.10.), потім збовтують у 200 мл знебарвлювального розчину протягом 15 хв і повторюють етап знебарвлювання щонайменше два чи три рази, доки середовище не стане прозорим і безбарвним. Потім споліскують пластину гелю дистильованою водою (2 × 2 хвилини) і висушують на повітрі (2-3 години) або феном (10-15 хвилин).
Примітка 1: Фіксування, промивання, забарвлювання і знебарвлювання здійснюють за температури 20 °С. Не допускають підвищених температур.
Примітка 2: Якщо перевагу надають більш чутливому методу забарвлювання сріблом (напр., Silver Staining Kit, Protein, Pharmacia Biotech, Code No 17-1150-01), оброблені плазміном казеїнові проби повинні бути розведені до 5 мг/мл.
Оцінювання проводять шляхом порівняння білкових смуг невідомої проби з контрольними стандартами на тому самому гелі. Виявлення коров’ячого молока у сирах з овечого молока, козячого молока і буйволячого молока та сумішей овечого, козячого і буйволячого молока здійснюють за допомогою γ3-казеїнів та γ2-казеїнів, ізоелектричні точки яких варіюються в діапазоні між pH 6,5 та pH 7,5 (рисунки 4 a, b, рисунок 5). Межа детектування становить менше ніж 0,5 %.
Для візуальної оцінки кількості коров’ячого молока рекомендується скоригувати концентрації проб та стандартів таким чином, щоб отримати такий самий рівень інтенсивності візуалізації овечих, козячих та/або буйволячих γ2-казеїнів та γз-казеїнів (див. ‘γ2 E,G,B’ та ‘γ3 E,G,B’ на рисунках 4 a, b та рисунку 5). Після цього кількість коров’ячого молока (менше ніж, дорівнює чи більше ніж 1 %) у невідомій пробі може бути оцінена безпосередньо шляхом порівняння інтенсивності візуалізації коров’ячих γ3-казеїнів та γ2-казеїнів (див. ‘γ3 С’ та ‘γ2 С’ на рисунках 4 a, b та рисунку 5) з тими у 0 % та 1 % контрольних стандартах (овечі та козячі), або внутрішньолабораторних стандартах (буйволячі).
За можливості, застосовують денситометрію (5.19.) для визначення співвідношення площі піків коров’ячих до овечих, козячих та/або буйволячих γ2-казеїнів та γ3-казеїнів (див. рисунок 5). Порівнюють це значення зі співвідношенням площі піків γ2-казеїнів та γ3-казеїнів 1 % контрольного стандарту (овечі та козячі) або внутрішньолабораторного стандарту (буйволячі), досліджуваних на тому самому гелі.
Примітка: Цей метод працює задовільно, якщо є чіткий позитивний сигнал стосовно обох коров’ячих γ2-казеїнів та γ3-казеїнів в 1 % контрольному стандарті, але не в 0 % контрольному стандарті. Якщо ні, процедуру оптимізують, точно дотримуючись детальних інструкцій методу.
Пробу оцінюють як позитивну, якщо обидва коров’ячі γ2-казеїни та γ3-казеїни або відповідні співвідношення площі піків дорівнюють або перевищують рівень 1 % контрольного стандарту.
8. ПЕРЕЛІК ВИКОРИСТАНОЇ ЛІТЕРАТУРИ
Addeo F., Moiо L., Chianese L., Stingo C., Resmini P., Berner I, Krause I., Di Luccia A., Восса A.: Use of plasmin to increase the sensitivity of the detection of bovine milk in ovine and/or caprine cheese by gel isoelectric focusing of γ2-caseins. Milchwissenschaft 45, 708-711 (1990).
Addeo F., Nicolai M.A., Chianese L., Moio L., Spagna Musso S., Восса A., Del Giovine L.: A control method to detect bovine milk in ewe and water buffalo cheese using immunoblotting. Milchwissenschaft 50, 83-85 (1995).
|
Krause I., Berner I, Klostermeyer H.: Sensitive detection of cow milk in ewe and goat milk and cheese by carrier ampholyte - and carrier ampholyte/immobilized pH gradient - isoelectric focusing of γ-caseins using plasmin as signal amplifier. in: Electrophoresis-Forum 89 (B.J. Radola, ed.) pp 389-393, Bode-Verlag, |
|
Krause I., Belitz H.-D., Kaiser К.-P.: Nachweis von Kuhmilch in Schaf and Ziegenmilch bzw. |
Radola B.J.: Ultrathin-layer isoelectric focusing in 50-100 μm polyacrylamide gels on silanised glass plates or polyester films. Electrophoresis 1, 43-56 (1980).
Схематичний малюнок покривного листа
Шар казеїну між водною та органічною фазами після центрифугування
Техніка «складання касети» для формування ультратонких поліакриламідних гелів
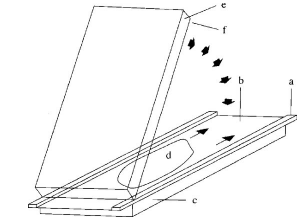
a = стрічка прокладка (0,25 mm); b = покривний лист (5.3.); c, e = скляні пластини (5.1.); d = розчин гелю (4.1.2); f = лист із нанесеним гелем (5.2.)
Ізоелектричне фокусування оброблених плазміном казеїнів сиру з овечого та козячого молока, що містить різні кількості коров’ячого молока.
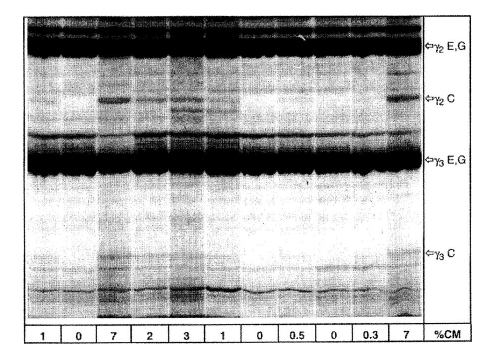
% СМ = відсотковий вміст коров’ячого молока, C = коров'яче, E = овече, G = козяче
Показана верхня частина IEF гелю.
Ізоелектричне фокусування оброблених плазміном казеїнів сиру, виготовленого із сумішей овечого, козячого та буйволячого молока, що містить різні кількості коров’ячого молока.
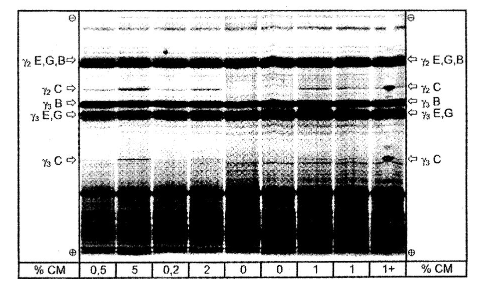
% СМ = відсотковий вміст коров’ячого молока; 1 + = проба, що містить 1 % коров'ячого молока та з доданим чистим коров’ячим казеїном на середині доріжки. C = коров'яче, E = овече, G = козяче, B = буйволяче.
Показано повну відстань розділення IEF гелю.
Суперпозиція денситограм стандартів (STD) та проб сиру, виготовленого із суміші овечого та козячого молока після ізоелектричного фокусування.
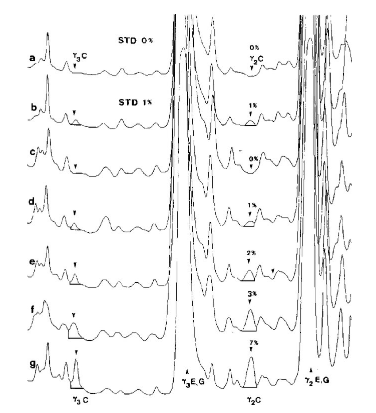
a, b = стандарти, що містять 0 та 1 % коров’ячого молока; c-g = проби сиру, що містять 0, 1, 2, 3 та 7 % коров'ячого молока. C = коров’яче, E = овече, G = козяче.
Відскановано верхню частину IEF гелю при λ = 634 нм.
Оцінювання результатів досліджень
Дослідження проводяться в лабораторіях, призначених відповідно до статті 12 Регламенту (ЄС) № 882/2004 (**), або призначених компетентними органами держави-члена.
2. Відбирання проб та спори щодо результатів досліджень
1. Відбирання проб проводиться згідно з нормативними положеннями, які стосуються обумовленого продукту. Якщо жодних положень щодо відбирання проб спеціально не передбачено, тоді застосовуються положення, визначені в ISO 707, Молоко та молочні продукти - Настанови щодо відбирання проб.
2. Лабораторні звіти про результати досліджень повинні містити інформацію, достатню для оцінювання результатів, яке має бути проведене згідно з доповненням.
3. Для досліджень, що вимагаються згідно з правилами Союзу, повинні бути відібрані дублікати проб.
4. Якщо виникає спір щодо результатів, платіжна агенція повинна забезпечити проведення необхідного дослідження обумовленого продукту повторно, а витрати повинна нести сторона, що програла.
Згадане вище дослідження проводиться за умови, що запечатані дублікати проб продукту є в наявності і належно зберігаються в компетентному органі. Виробник повинен надіслати запит платіжній агенції на проведення дослідження протягом 7 робочих днів після нотифікації результатів першого дослідження. Дослідження повинна проводити платіжна агенція протягом 21 робочих днів після отримання запиту.
5. Результат повторного дослідження є остаточним.
6. Якщо виробник може довести, протягом п’яти робочих днів з моменту відбирання проб, що процедура відбирання проб не була проведена коректно, відбирання проб повинно бути повторене, якщо це можливо. Якщо відбирання проб не може бути повторене, партія повинна бути прийнята.
Оцінювання партії на відповідність нормативно встановленій межі (допустимим концентраціям)
Якщо законодавство стосовно державних інтервенцій та допомоги на зберігання на складах приватного користування встановлює детальні процедури відбирання проб, тоді слід дотримуватись тих процедур. У всіх інших випадках використовується проба із щонайменше 3-х одиничних проб, довільно відібраних з партії, поданої для контролю. Допускається підготування змішаних проб. Отриманий результат порівнюється з допустимими концентраціями шляхом обчислення 95 % довірчого інтервалу як помноженого на 2 стандартного відхилу, де стандартний відхил залежить від того, чи (1) метод валідований у рамках міжнародної співпраці з визначеними величинами дляσг та σR, або чи (2), у випадку внутрішньої валідації, була обчислена внутрішня відтворюваність. Цей довірчий інтервал тоді дорівнюватиме невизначеності вимірювання результату.
2. Метод валідований у рамках міжнародної співпраці
У цьому випадку, стандартний відхил повторюваності σг та стандартний відхил відтворюваності σR встановлені, і лабораторія може підтвердити відповідність робочим характеристикам валідованого методу.
Обчислюють середнє арифметичне Формула числа n повторених вимірювань.
Обчислюють розширену невизначеність (k = 2) для Формула за формулою
Якщо кінцевий результат х вимірювання обчислюють з використанням формули типу х = у1 + у2, х = у1 - у2, х = у1 • у2 або х = у1/y2, у таких випадках дотримуються звичайних процедур об’єднання стандартних відхилів.
Партію вважають такою, що не відповідає верхній межі допустимої концентрації UL, якщо
в іншому випадку, її вважають такою, що відповідає UL.
Партію вважають такою, що не відповідає нижній межі допустимої концентрації LL, якщо
в іншому випадку, її вважають такою, що відповідає LL.
3. Внутрішня валідація з обчисленням стандартного відхилу внутрішньої відтворюваності
У випадках, коли використовують методи, не зазначені в цьому Регламенті, а міри прецизійності не були встановлені, проводиться внутрішня валідація. У формулах для обчислення розширеної невизначеності U використовуються стандартний відхил внутрішньої повторюваності σіг та стандартний відхил внутрішньої відтворюваності σiR замість σг та σR , відповідно.
Правила, яких слід дотримуватись перевірки на відповідність допустимій концентрації, зазначені в пункті 1. Однак, якщо партію вважають такою, що не відповідає допустимій концентрації, вимірювання повинні бути повторені з використанням методу, зазначеному в цьому Регламенті, а результати оцінені згідно з пунктом 1.
(*) Продукти Ampholine® pH 3,5-9,5 (Pharmacia) та Resolyte® pH 5-7 і pH 6-8 (BDH, Merck) виявились особливо придатними для отримання потрібного розділення γ-казеїнів.
(**) Регламент Європейського Парламенту і Ради (ЄС) № 882/2004 від 29 квітня 2004 року про офіційний контроль для забезпечення перевірки на відповідність законодавству щодо кормів та харчових продуктів, правил щодо здоров'я та благополуччя тварин (OB L 165, 30.04.2004, с. 1).
__________
(-1) Метод, який застосовуватимуть, повинен бути схвалений платіжною агенцією.
(-2) Дослідження пригорілих частинок можуть проводитись систематично. Проте, такі дослідження повинні завжди проводитись, якщо не здійснюються органолептичні дослідження.
(-3) Метод, який застосовуватимуть, повинен бути схвалений платіжною агенцією (один або обидва методи).
(-4) Метод, який застосовуватимуть, повинен бути схвалений платіжною агенцією.
(-5) Органолептичні дослідження повинні здійснюватися, якщо це вважають за необхідне, після аналізу ризику, схваленого платіжною агенцією.
(-6) Метод, який застосовуватимуть, повинен бути схвалений платіжною агенцією.
(-7) Нанесення проби: Після попереднього фокусування (етап 1), наносять дозатором 18 мкл проби та стандартних розчинів на аплікатори проб (10 × 5 мм), кладуть їх на гель на відстані 1 мм один від одного та 5 мм повздовжньо від анода і легко притискають. Здійснюють фокусування за зазначених вище умов, обережно знімають аплікатори проб після 60 хвилин фокусування проби.
{Джерело: Урядовий портал (Переклади актів acquis ЄС) https://www.kmu.gov.ua}